Home » Health News »
A full model of the SARS-CoV-2 spike protein for studying its atomic-level dynamics by multiple microsecond simulations
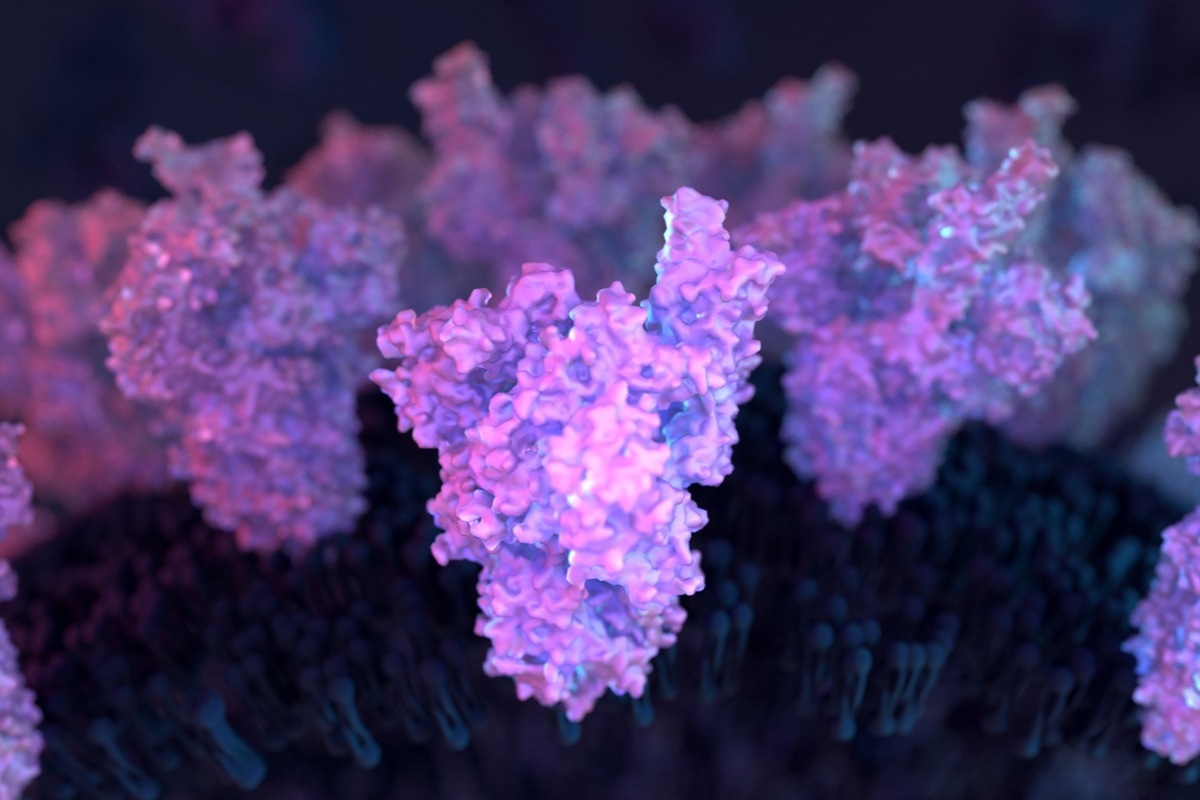
In a recent study published in the PNAS, researchers constructed a full-length severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S) structure in its membrane-bound state to study functionally relevant aspects of its global conformational dynamics.
Background
SARS-CoV-2 S is a glycoprotein with all necessary components that facilitate its entry into human cells (host). Briefly, it binds to the surface receptors, such as angiotensin-converting enzyme 2 (ACE2) on the human cell membrane, initiates the fusion of two membranes, and finally releases the viral genome into the host cell.
Cryo-electron microscopy (cryo-EM) has resolved several S structures; however, essential structural details of the globular domain of SARS-CoV-2 S (S head) are missing. Cryo-EM structures also lack critical information about the conserved fusion peptide (FP), stalk heptad repeat 2 (HR2) domain, and the transmembrane (TM) ectodomain. Likewise, the information about the glycosylation of different S residues is missing.
These regions of SARS-CoV-2 S are conserved between coronaviruses (CoVs), while the TM domain contains multiple palmitoylation sites that stabilize S in the envelope, glycosylation of the S residues is crucial for the S protein folding, shaping viral tropism, and shielding SARS-CoV-2 from immune recognition. Additionally, structural studies have not accounted for the cellular environment where SARS-CoV-2 assembles before budding and releasing infectious virions, i.e., the endoplasmic reticulum–Golgi intermediate compartment (ERGIC).
Overall, a complete picture of the global conformational dynamics of the S is non-existent. Advanced studies deploying molecular dynamics (MD) simulations have revealed critical information about several functionally relevant features of S. For instance, these studies explored how glycans shield the S from the immune system or how the S head facilitates the receptor-binding domain (RBD) to switch between its active (up) and inactive (down) conformations.
Yet, scientists do not know how glycosylation contributes to S protein’s global conformational dynamics or the role of palmitoylations in the TM domain. In a nutshell, characterizing the mechanism of SARS-CoV-2 S action remains critical for understanding how it establishes infection and developing therapeutic strategies against SARS-CoV-2.
About the study
In the present study, researchers used micro-second scale MD simulations to study the motion of the S head with respect to the stalk. In addition, they critically investigated the roles of glycan–lipid and glycan–glycan interactions in the bending and twisting motions of the S head. Finally, the researchers simulated palmitoylation effects in the TM domain of the S head. Notably, S-palmitoylation is a covalent lipid modification of the Cysteine residues that modulates the host membrane curvature.
The hybrid approach taken by the researchers combined protein-protein docking, homology modeling, and the available experimental data. They first validated the modeled parts of the S protein and then used MD simulations to characterize its global conformational dynamics, focusing on the glycans.
Study findings
This study provided insights into several functionally relevant features of S protein’s global conformational dynamics. First, it demonstrated that bending and twisting motions of the S head gave a high degree of flexibility to the RBD. Hence, it effectively located and bound the ACE2 receptors in the host cells. Bending and twisting motions of the S head around the stalk were quantified in terms of head-twist and head-bend angles, as well as S head distances calculated separately with respect to the S neck, the HR2 domain, or the TM ectodomain.
The SARS-CoV-2 S head (in fully-glycosylated form) showed the highest conformational flexibility, especially in head-twist angles along its HR2 and TM domains. The highest sampled configurations turned by almost 90° relative to the starting structure. Conversely, a comparatively smaller range of motion was allowed around the S neck. These results could be implicated in the length of the linkers connecting different S domains. In the non-glycosylated system, these motions became surprisingly restricted, indicating the S dynamics accentuating the role of glycans. Similarly, the fully-glycosylated S head moved as much as 125 Å with respect to the HR2 domain; moreover, the S domains displayed a relatively smaller range of motion in the absence of glycans.
The researchers identified reasons for the flexibility of linkers or hinge regions. These three linkers connected the S head with S neck (NTVYDPL), S neck with HR2 domain (HTSPDVDLGDISG), and HR2 domain with TM domain (GKYEQYIKWP). They found that these linkers were rich in Proline (Pro) and Glycine (Gly) amino acid residues which facilitated the hinge or recoil motion contributing to the conformational flexibility of S. All the Pro and Gly residues present in the flexible regions are conserved between human SARS-CoV and SARS-CoV-2 giving an evolutionary advantage. Also, high glycan densities were observed around these flexible regions and the stalk, providing a shield for the SARS-CoV-2 S against the immune response and neutralizing antibodies.
Furthermore, MD simulations revealed that the glycans formed contact with each other and the lipid bilayer. The authors also observed a correlation between the bending of the HR2 domain and the glycan interactions. In the absence of glycan–lipid interactions, the HR2 domain could move freely only within the 20° inclination. They observed that in the range of 20 to 50 Å from the center of the TM domain, the palmitoylated TM domain was found within a positively curved membrane, indicating SARS-CoV-2 S palmitoylation contributed to the bending of the host cell membrane.
The correlated interdomain motions of SARS-CoV-2 S were attributable to its unique tertiary structure. However, glycosylation further enhanced these motions through the extensive glycan–lipid and glycan–glycan interactions. The magnitude of S interdomain motion and correlation between them reduced in the absence of glycosylation. Subsequently, the researchers observed a lower Pearson’s coefficient, 0.76 ±0.07, for the non-glycosylated S.
- Posttranslational modifications optimize the ability of SARS-CoV-2 spike for effective interaction with host cell receptors. Karan Kapoor, et al. PNAS. doi: https://doi.org/10.1073/pnas.2119761119 https://www.pnas.org/doi/full/10.1073/pnas.2119761119
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: ACE2, Amino Acid, Angiotensin, Angiotensin-Converting Enzyme 2, Antibodies, Cell, Cell Membrane, Coronavirus, Coronavirus Disease COVID-19, Cysteine, Electron, Electron Microscopy, Enzyme, Genome, Glycan, Glycans, Glycine, Glycoprotein, Glycosylation, Immune Response, Immune System, Membrane, micro, Microscopy, Neck, Proline, Protein, Protein Folding, Receptor, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Spike Protein, Syndrome

Written by
Neha Mathur
Neha is a digital marketing professional based in Gurugram, India. She has a Master’s degree from the University of Rajasthan with a specialization in Biotechnology in 2008. She has experience in pre-clinical research as part of her research project in The Department of Toxicology at the prestigious Central Drug Research Institute (CDRI), Lucknow, India. She also holds a certification in C++ programming.
Source: Read Full Article


