Home » Health News »
Examining the causal mechanism behind rare hereditary diseases
 of a human cell. The HMGB1 protein (green) that is mutated in BPTA syndrome forms a hard layer at the nucleolus (pink), causing the developmental disorder. Credit: MPIMG | Henri Niskanen")
Universitätsmedizin Berlin, the Max Planck Institute for Molecular Genetics (MPIMG), and the University Hospital Schleswig-Holstein (UKSH) have investigated in detail how BPTA syndrome, an extremely rare hereditary condition, arises. A change in the charge of a protein disrupts cellular self-organization, resulting in a developmental disorder.
The team also identified hundreds of comparable genetic changes associated with various conditions, such as brain development disorders and predisposition to cancer. This mechanism, which has now been described in the journal Nature, could be the cause of numerous unexplained diseases and health conditions.
Thousands of genetic changes are associated with various diseases, disorders, and conditions. But how, exactly, these mutations produce disease is seldom clear. This is because the changes relate to sections of proteins with a disordered three-dimensional structure and a function inside the cell about which little is known so far.
“It’s hard to study what these kinds of protein segments are responsible for doing because in many cases, they have to interact with other molecules before producing their effects,” says Dr. Martin Mensah of the Institute of Medical and Human Genetics at Charité. He is one of the study’s two first authors and a fellow in the Digital Clinician Scientist Program, which Charité operates with the Berlin Institute of Health (BIH) at Charité.
“Taking BPTA syndrome as an example, we have now described in detail how changes in disordered areas of proteins can cause a genetic disease.” This means the research team has discovered a new mechanism that causes hereditary diseases– and, according to the study, one that is surprisingly not all that rare after all.
BPTA stands for “brachyphalangy, polydactyly and tibial aplasia/hypoplasia.” Patients have severe malformations affecting the extremities, face, nervous system and bones, and other organs. There are fewer than ten documented cases worldwide, making this disease extremely rare.
To identify the cause of this syndrome, the researchers decoded the genetic information of five of the affected patients and discovered a change in the HMGB1 protein in all of them. Due to what is known as a frameshift mutation, the final one-third of the protein’s structure has a positive charge rather than the usual negative one.
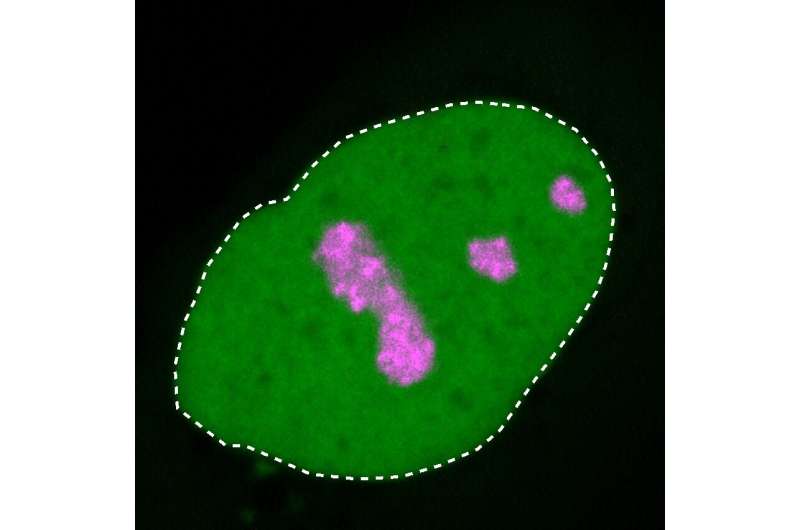
The nucleolus solidifies
The change in the charge means that HMGB1 resembles proteins that tend to cluster in the nucleolus, a small area in the nucleus of the cell where parts of the cell’s protein factories are assembled.
This role makes the nucleolus crucial to a cell’s viability. As the research team showed based on experiments on isolated proteins and cell cultures, the mutated HMGB1 protein, which now has a positively charged end section, is improperly drawn toward the nucleolus. And because the extension of the protein has also grown stiffer, the HMGB1 protein also clumps together.
“Under a microscope, we were able to see that this causes the nucleolus to lose its own liquid-like properties and become increasingly rigid,” explains Dr. Henri Niskanen, a researcher at MPIMG and the study’s other first author.
This solidification of the nucleolus adversely affects the cells’ vital functioning. More cells with the mutated protein than without the mutation died in the culture. Prof. Dr. Malte Spielmann, Director of the Institute of Human Genetics at UKSH and one of the study’s three lead authors, offers his conclusion: “We showed how mutations in disordered sections of proteins can cause a disease. When there is a change in the charge, the protein wrongly accumulates in the nucleolus, adversely affecting its vital functioning. This leads to a disorder in the organism’s development.”
New explanations for existing diseases
Following their initial findings, the researchers searched databases containing the DNA sequences of thousands of people, looking for similar cases. They were able to identify more than 600 mutations in 66 proteins that gave the final part of the protein both a positive charge and stiffer properties. Of those mutations, 101 had previously been associated with various diseases, including neurodevelopmental disorders and increased susceptibility to cancer.
For 13 selected proteins, the team studied the cell culture to see whether the mutations gave them a particular affinity for the nucleolus. That was the case for 12 of them. About half of the proteins tested impaired the functioning of the nucleolus, so they were similar to the disease mechanism found for BPTA syndrome.
“The mechanism that causes this disease, which we discovered in BPTA syndrome, could be implicated in many other diseases and conditions as well,” says Prof. Dr. Denise Horn, lead author of the study who works at the Institute of Medical and Human Genetics at Charité. “So we’ve opened a door that could help to explain many other diseases. The real work starts now.”
The newly identified mechanism could also lead to new therapeutic approaches, at least for some diseases. “Tumors are attributable to genetic changes in the affected cells,” explains Dr. Denes Hnisz, the head of a research group at MPIMG and the study’s third lead author. “This means we may be able to prevent cancer from developing in the future by intervening in the cell’s self-organization, which is mediated by disordered sections of proteins.”
More information:
Denes Hnisz, Aberrant phase separation and nucleolar dysfunction in rare genetic diseases, Nature (2023). DOI: 10.1038/s41586-022-05682-1. www.nature.com/articles/s41586-022-05682-1
Journal information:
Nature
Source: Read Full Article

